罕见遗传病科普——杜氏肌营养不良症
在满心期待中,小明出生了,大家喜出望外,奔走相告,家里添了一名男丁啦!
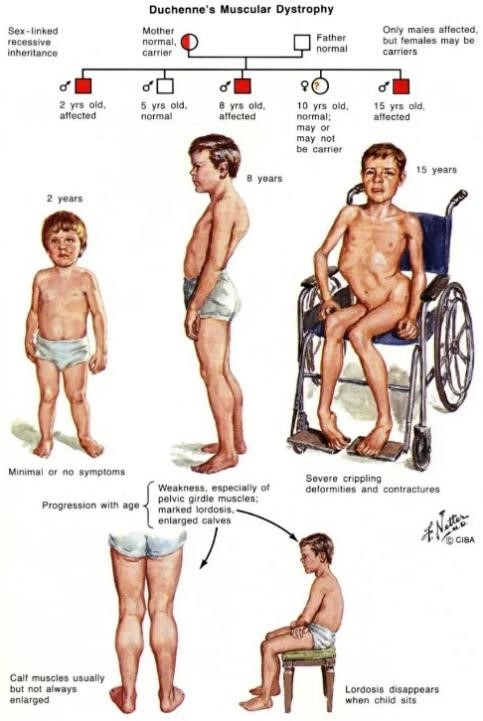
然而随着时间的流逝,幸福戛然而止,他不会走路了;
他走路晚,易摔倒,走路慢
他3岁左右出现明显的步态异常,跑跳、爬楼等运动逐渐出现困难,双侧腓肠肌假性肥大
他4-5岁运动能力开始倒退
他10-12岁丧失独立行走的能力,需借助轮椅行动
他成绩差,智力异常
(图片来自网络)
这是得了什么病,怎么突变不能走路了,智力也出现了异常?
今天我们来认识一个罕见的遗传病——杜氏肌营养不良症(Duchenne muscular dystrophy, DMD),以下简称DMD。
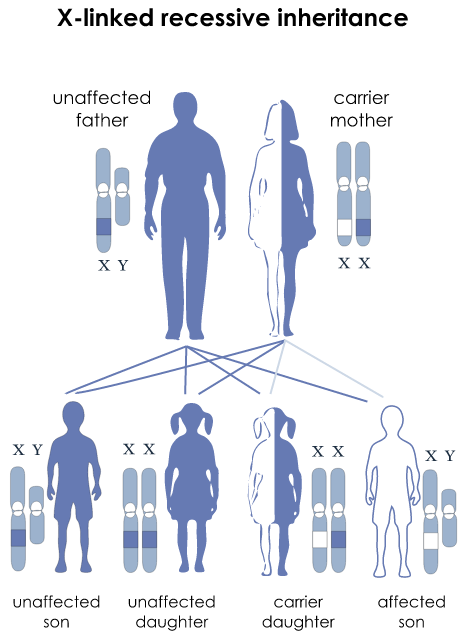
DMD是一种X-连锁隐性遗传性神经肌肉病
(图片来自网络)
1、患者几乎全为男性,活产男婴发病率:1/3500
2、女性多为致病基因携带者,一般不会表现出症状。女性患者极少,这可能与X染色体失活相关。有症状的女性携带者1/100,000-1/450,000
3、主要临床表现:进行性、对称性肌无力,近端重于远端,伴有双侧腓骨肌假性肥大
4、致病基因:DMD
5、神经肌电图:肌源性损害
6、肌酶谱:患者CK、LD、CK-MB通常有异常显著增高(正常值的20-100倍),但晚期可明显下降;约50%携带者CK可轻度增高
7、肌活检: 免疫组化可见几乎无抗肌萎缩蛋白(正常对照的0-5%)
致病基因
DMD基因,是目前已知人类最大的基因,位于Xp21.2,全长约2.5 Mb,cDNA含79个外显子,包括多个独立组织特异性的启动子和2 个PolyA 加尾位点。
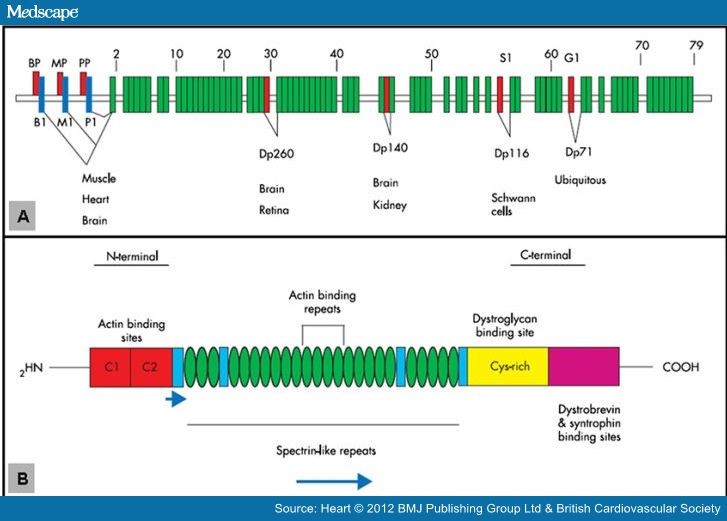
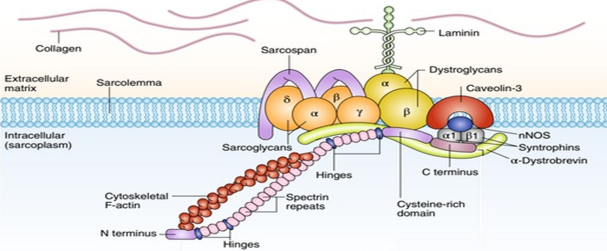
DMD基因编码dystrophin蛋白(抗肌萎缩蛋白),dystrophin主要分布于骨骼肌和心肌细胞,通过其N端和C端分别与细胞骨架和细胞膜,起支架作用,可以保护肌细胞膜在肌肉收缩时不受损伤。其缺乏可破坏肌细胞膜的完整性,出现异常蛋白和钙内流、肌酸激酶外流,最终造成肌细胞变性、坏死。
变异类型
1、 DMD基因变异约60%为外显子缺失,缺失热点为2-13和44-52号外显子。
2、 10%为DMD基因外显子重复,无明显热点。
3、约30%为其他类型突变,包括点突变、小的插入/缺失突变、少量患者可能与编码区外突变有关。
遗传学诊断方法
1、多重连接探针扩增法(MLPA):检测79个外显子的重复或者缺失;
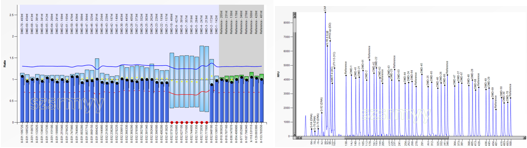
2、DNA测序:可明确DMD基因的点突变和微小变异;
治疗方法
1、基因治疗
1)、外显子跳跃策略,如2016年FDA批准上市的反义寡核苷酸药物Eteplirsen(商品名Exondys 51),可诱导mRNA跳过51外显子,生成部分缺失但具有功能的蛋白,可治疗开放读码框被破坏的患者(约占15%)。
2)、终止密码子通读策略,如2014年欧盟委员会批准上市的Ataluren(商品名Translarna),可减少核糖体对提前终止密码子的敏感性,使mRNA翻译时不在终止密码子处停止,可治疗DMD无义突变患者(约占13%)。
3)、外源性微小抗肌萎缩蛋白基因替代策略。
4)、基因修复治疗策略等。
2、支持治疗
糖皮质激素、营养管理、康复训练、无创通气、机械通气、脊柱矫形器、手术等。
临床咨询
1、患者父亲亲通常不是携带者,也不会有症状,家系中存在2个或以上的患者,患者的母亲通常是携带者。
2、家系中有1个患者,则患者母亲可能是携带者,也可能不是。需要注意的是,即使外周血检测未发现患者母亲携带与患者相同的致病变异,其仍可能由于生殖腺嵌合(15%~20%)再次生育患病的后代。
3、对于 DMD 患儿的母亲,无论其是否为携带者,在其再次生育时均应进行产前诊断。
残余风险
综合应用 MLPA 和基因测序,仍有约6.9%的患者找不到致病变异。
应考虑以下的可能性:
1、 突变位于内含子深处或表达调控区,此时可通过 RT - PCR 方法直接检测患者 DMD 基因的表达水平;
2、患者的临床诊断不准确,需重新进行详细的临床评估,必要时进行肌活检。
【收藏此页】 【打印本页】 【关闭窗口】
 官方微信
官方微信




