关爱SMA儿童,关注SMA疾病
自1996年起,包括欧洲、美国、加拿大、澳大利亚等地在内的全球SMA患者群体将每年的8月定为SMA关爱月,提升社会公众对SMA这一罕见疾病的了解,以及对SMA群体的关爱与支持。在2022年8月7日已是第5个“国际脊髓型肌萎缩症(SMA)关爱日”。
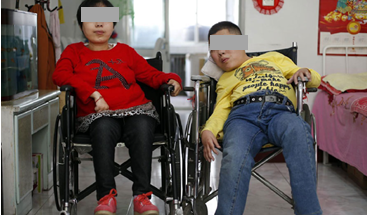
什么是SMA?
SMA为脊髓性肌萎缩症( spinal muscular atrophy,SMA) ,是导致 2 岁以下儿童死亡原因中最常见的遗传性疾病,有 80%的Ⅰ型患儿会在1岁内死亡,很少活过2岁。
l 一种常染色体隐性遗传病,夫妻双方为携带者,则有25%的几率诞下患儿。
l 运动神经元存活基因 1(SMN1)变异导致 SMN 蛋白功能缺陷所致的遗传性神经肌肉病
l SMA 以脊髓前角运动神经元退化变性和丢失导致的肌无力和肌萎缩为主要临床特征
l 临床表现为:进行性、对称性肌无力和肌萎缩,近端重于远端,下肢重于上肢
l 智力正常
为什么需要关注SMA?
l SMA是罕见病中的常见病
虽然新生儿中, 10000个只有1个SMA患儿,但是普通人群中,SMA致病基因携带率非常高,达到40-50人就有一个是SMA携带者。据统计,我国约有3000万SMA携带者,一旦夫妻双方同为携带者,则每一胎均有25%的概率生育SMA患儿
l SMA严重影响患者寿命及生存质量
根据起病年龄、运动里程碑及病情进展程度,SMA可分为五型
表型 | 占比 | 发病年龄 | 存活时间 | 最大运动能力 | 临床表现 |
SMA 0 | 很少 | 出生前、出生时 | 数月 | 无 | 除眼球外,几乎无肢体、躯干和面部的任何活动,无吸吮动作; 先天性关节挛缩、肌肉萎缩、反射消失; 出生后即需要呼吸机辅助呼吸; 可合并先天性心脏病 |
SMA I | 40-50% | <6个月 | 通常≤2 岁,随着改善呼吸和营养治疗,也可存活更久 | 需要扶坐 | 松软儿,严重肌张力低下,四肢无力; 舌肌、面肌、咀嚼肌无力; 胸廓钟型; 容易反复呼吸道感染及呼吸衰竭 |
SMA II | 30-40% | 6-18个月 | 大部分可存活至成年 | 可独坐,不能独立行走 | 婴幼儿期出现缓慢加重的全身性肌无力和肌张力低下; 运动发育落后,舌肌纤颤或手部肌束颤; 可伴关节挛缩及脊柱侧弯,影响呼吸功能 |
SMA III | 10-20% | >18个月 | 寿命不缩短或轻度下降 | 可独立行走 | 生后1年内运动发育正常; 儿童期逐渐出现近端为主的肌无力,下肢重于上肢; 疾病进展丧失行走能力; 可见肌束颤;后期出现脊柱侧弯、关节畸形、呼吸功能不全等表现 |
SMA IV | 很少 | 成人(常20-30岁) | 寿命一般不受影响 | 能跑跳等所有运动能力 | 青少年期或成人期起病,下肢起始的四肢近端无力,病情缓慢进展 |
SMA 0最严重出生前、出生时即发病,只能存活数月,除眼球外,几乎无肢体、躯干和面部的任何活动;
SMA I最常见,在6个月前发病,患者永远无法坐下或行走,通常会在2岁前死亡。
SMA IV最少见,症状最轻,能跑跳等所有运动能力,寿命一般不受影响。
l SMA治疗极其昂贵
目前有3钟上市药物,可以改善SMA患者的生存质量及存活时间,但是价格极其昂贵:
Nusinersen(商品名诺西那生钠Spinraza),每剂费用为 125,000 美元,在多次给药的过程中,其治疗费用将超过100万美元;
Onasemnogene abeparvovec-xioi被称为“世界最昂贵的药”,一针费用高达210万美元,相当于1000多万人民币
isdiplam 的价格为每年 340,000 美元
2021年12月3日,国家医疗保障局将全球首个SMA(脊髓性肌萎缩症)治疗药物诺西那生钠注射液正式纳入医保。将注射液的价格从曾经的一针约70万元,大幅降价至现在的3万多元,极大降低了患者及家庭负担,为SMA患者群体带来了新希望。
SMA如何预防?
l SMA致病机制
SMA是由于运动神经元存活基因 1(SMN1)变异导致 SMN 蛋白功能缺陷所致的遗传性神经肌肉病。其中SMN1基因决定疾病的发生,SMN2基因影响疾病的严重程度和进展 。
SMN1基因,为SMA致病基因,位于5q11.2-q13.3,全长约20kb,100%编码脊髓运动神经元存活蛋白。
约95%SMA患者由SMN1双等位基因纯合突变所致,即[0+0]基因型,大部分为外显子7和8共同缺失,少部分为外显子7。(外显子8为 非编码区,SMN1缺失通常指外显子7缺失)
5%由SMN1复合杂合突变所致,即一个等位基因缺失,另一个等位基因 发生微小致病突变,为[0+1d]基因型
SMN1双等位基因均发生微小致病突变,即[1d+1d]基因型,非常罕见
SMN2基因,与SMN1基因毗邻且高度同源,仅有5个核苷酸的差异。其中在第7外显子第6位c.840C/T,导致90%的SMN2基因mRNA外显子7被选择性剪接,仅有10%的SMN2表达全长有功能SMN蛋白,患者携带SMN2拷贝数越多表型越轻。
l SMA携带者筛查
SMN1基因检测进行携带者筛查是目前最好的预防手段。了解了SMA的致病基因,对其携带者筛查,是预防SMA患儿出生最有效最简单的方法。欧美等西方国家早已将SMA作为人群普筛的公共卫生项目,以降低SMA患儿的出生。2008年美国对所有孕龄人群无差别实施SMN1携带者基因筛查,对高风险胎儿实施产前诊断,从而减少SMA患儿的出生。
l SMA筛查及诊断对象
普通人群携带者筛查:
孕前体检人群,产前检查(早、中孕期)孕妇。
新生儿筛查
对新生儿进行早筛查,实现早发现、早诊治,避免或减少危害
产前诊断对象:
1. 生育过SMA患儿的夫妻;
2. 夫妻双方均为SMA携带者;
3. 生育过临床诊断SMA患儿,但患儿夭折前未进行基因诊断,再生育前通过SMN1检测明确双方是否为SMA携带者 SMA患者及家系诊断分析疑似SMA患儿、生育过SMA患儿或者家族中出现过类似症状亲属的家庭,可以进行SMA的基因诊断,以明确是否患病及其类型,以便采取后续治疗措施或制定生育计划。
深圳市人民医院法医物证室,自2018年开始,便常规开展了外周血/羊水/脐血的SMN1 基因杂合缺失携带者筛查、SMA患者的基因诊断等项目。完善了SMA检测前咨询、实验样本检测、报告解读、及后续产前诊断的规范化流程,迄今为止已检测了约八千例样本,成功地帮助了SMA患儿家庭生育健康的孩子。法医物证室致力于SMA基因检测技术的开发和临床应用,参与完成了2项SMA基因检测试剂盒的临床试验(GCP)。积极参加SMA学术沙龙,与专家同行探讨SMA的临床检测技术及应用,普及和发展深圳市SMA携带者筛查项目的,有效地降低了中国家庭生育SMA患儿的风险。

总之,早预防、早干预是应对SMA经济有效的办法。深圳市人民医院法医物证室可进行SMA携带者筛查、产前诊断、SMA患者及家系诊断 ,分析下一代的患病风险,有效预防新生儿出生缺陷的发生,为深圳市妇女儿童的健康平安保驾护航。
【收藏此页】 【打印本页】 【关闭窗口】
 官方微信
官方微信




